

kromatografi, også kendt som "kromatografisk analyse", "kromatografi", er en separations- og analysemetode, som har en meget bred vifte af anvendelser inden for analytisk kemi, organisk kemi, biokemi og andre områder.
Grundlæggeren af kromatografi er en russisk botaniker M.Tsvetter.I 1906 offentliggjorde den russiske botaniker Zvetter resultaterne af sit eksperiment: For at adskille plantepigmenter hældte han petroleumsetherekstrakt indeholdende plantepigmenter i et glasrør indeholdende calciumcarbonatpulver og eluerede det med petroleumsether fra top til bund.Fordi forskellige pigmenter har forskellige adsorptionskapaciteter på overfladen af calciumcarbonatpartikler, med udvaskningsprocessen, bevæger forskellige pigmenter sig ned med forskellige hastigheder og danner således bånd af forskellige farver.Pigmentkomponenterne blev adskilt.Han kaldte denne separationsmetode kromatografi.
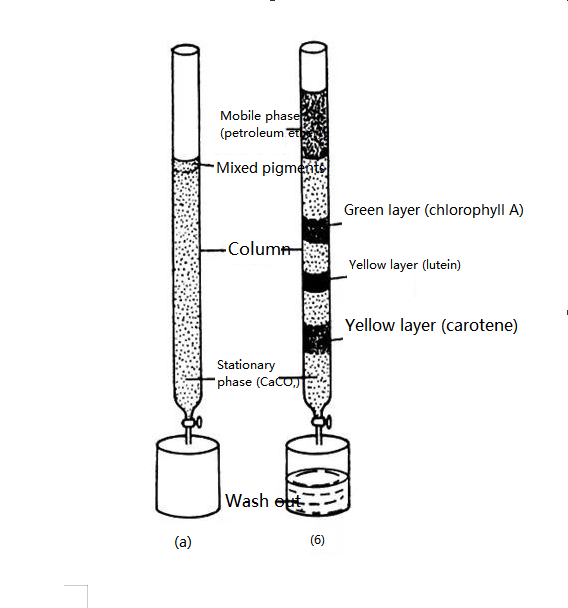
Skematisk fremstilling af et plantebladspigmentadskillelseseksperiment
Med den fortsatte udvikling af separationsmetoder bliver flere og flere farveløse stoffer genstand for separation, kromatografi mistede også gradvist betydningen af "farve", men navnet er stadig i brug i dag.
Kromatografisk klassificering
Essensen af kromatografi er en proces, hvor molekylerne, der skal adskilles, opdeles og balanceres mellem den stationære fase og den mobile fase.Forskellige stoffer er fordelt forskelligt mellem de to faser, hvilket får dem til at bevæge sig med forskellig hastighed med den mobile fase.Med bevægelsen af den mobile fase adskilles forskellige komponenter i blandingen fra hinanden på den stationære fase.Afhængigt af mekanismen kan den opdeles i en række kategorier.
1, ifølge den tofasede fysiske tilstandsklassifikation
Mobil fase: Gaschromatografi, væskekromatografi, superkritisk væskekromatografi
Stationær fase: gas-fast, gas-væske;Væske-fast, væske-væske
2, i henhold til formen for stationær faseklassificering
Søjlekromatografi: pakkesøjlekromatografi, kapillarsøjlekromatografi, mikropakket søjlekromatografi, præparativ kromatografi
Plankromatografi: papirkromatografi, tyndtlagskromatografi, polymermembrankromatografi
3, klassificeret i henhold til separationsmekanismen
Adsorptionskromatografi: Forskellige komponenter adskilles efter deres adsorptions- og desorptionskapacitet på adsorbenter
Fordelingskromatografi: De forskellige komponenter adskilles efter deres opløselighed i opløsningsmidlet
Molekylær udelukkelseskromatografi: ifølge størrelsen af molekylstørrelsen af separationen ln ionbytterkromatografi: forskellige komponenter af affiniteten for ionbytterharpiksseparationen
Affinitetskromatografi: Separation ved hjælp af tilstedeværelsen af en specifik affinitet mellem biologiske makromolekyler
Kapillærelektroforese: komponenterne blev adskilt i overensstemmelse med forskellene i mobilitet og/eller partitionsadfærd
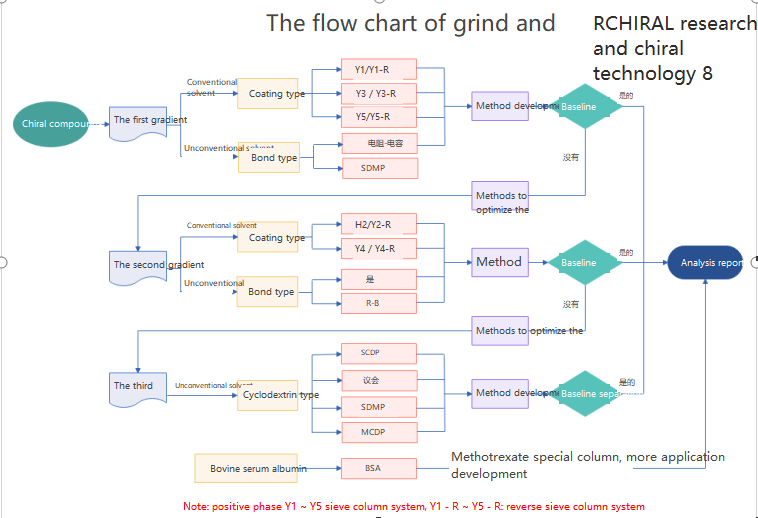
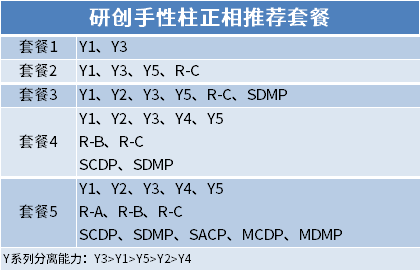
Chiral kromatografi bruges til adskillelse og analyse af chirale lægemidler, som kan opdeles i tre kategorier: chiral derivatiseringsreagensmetode;Kiral mobil fase additiv metode;Kiral stationær faseopløsningsmetode
Grundlæggende terminologi for kromatografi
Kurverne opnået ved at plotte komponenternes responssignaler efter påvisning af kromatografisk adskillelse mod tid kaldes kromatogrammer.
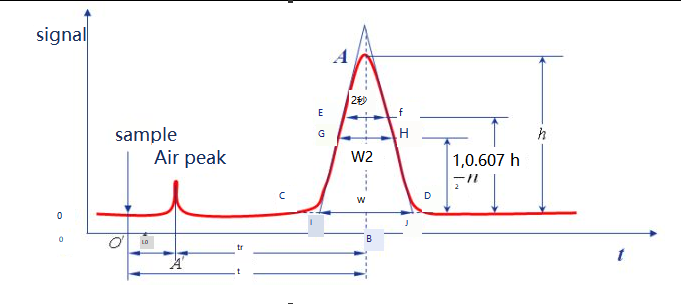
Baseline:Under visse kromatografiske forhold kaldes kurven for signalet, der genereres, når kun den mobile fase passerer gennem detektorsystemet, basislinjen, som vist i ot-linjen.Når den eksperimentelle tilstand var stabil, var basislinjen en linje parallel med den vandrette akse.Basislinjen afspejler instrumentets støj, primært detektoren, over tid.
Tophøjde:den lodrette afstand mellem det kromatografiske toppunkt og basislinjen, angivet med h, som vist i AB'-linjen.
Regionsbredde:Regionsbredden af den kromatografiske top er direkte relateret til separationseffektiviteten.Der er tre metoder til at beskrive kromatografisk spidsbredde: standardafvigelse σ, spidsbredde W og FWHM W1/2.
Standardafvigelse (σ):σ er den halve afstand mellem de to bøjningspunkter på normalfordelingskurven, og værdien af σ angiver graden af spredning af komponenterne væk fra søjlen.Jo større værdien af σ er, jo mere spredt er spildevandskomponenterne, og jo værre er separationseffekten.Omvendt er spildevandskomponenterne koncentrerede, og separationseffekten er god.
Spidsbredde W:Skæringspunkterne på begge sider af den kromatografiske top bruges som tangentlinjer, og skæringspunktet på basislinjen kaldes peak width, eller baseline width, som også kan udtrykkes som W, som vist i figur IJ.Ifølge normalfordelingsprincippet kan sammenhængen mellem spidsbredde og standardafvigelse bevises at være W=4σ.
W1/2:Topbredden ved halvdelen af tophøjden kaldes FWHM, som vist for afstanden af GH.W1/2=2,355σ, W=1,699W1/2.
W1/2, W er begge afledt af σ og bruges til at beregne toparealer ud over at måle søjleeffekten.FWHM-måling er mere praktisk og mest almindeligt anvendt.
kort opsummering
Fra den kromatografiske topudstrømningskurve kan følgende mål opnås:
a, Kvalitativ analyse blev udført baseret på retentionsværdien af kromatografiske toppe
b, kvantitativ analyse baseret på arealet eller toppen af den kromatografiske top
C. Separationseffektiviteten af søjlen blev evalueret i overensstemmelse med retentionsværdien og topbredden af den kromatografiske top
Beregningsformlen involveret i kromatografi
1. Retentionsværdi
Retentionsværdien er en parameter, der bruges til at beskrive, i hvilken grad en prøvekomponent tilbageholdes i kolonnen og bruges som en indikator for kromatografisk karakterisering.Dens repræsentationsmetode er som følger:
Retentionstid tR
DødstidspunktettM
Juster retentionstiden tR'=tR-tM
(Samlet tid brugt i stationær fase)
Retentionsvolumen
VR=tR*F. (uafhængig af mobilfasehastighed)
Død volumen
VM=tM*Fc
(Den plads, der ikke er optaget af den stationære fase i strømningsvejen fra injektoren til detektoren)
Juster retentionsvolumen VR'=t'R*Fc
2. Relativ fastholdelsesværdi
Relativ retentionsværdi, også kendt som separationsfaktor, fordelingskoefficientforhold eller relativ kapacitetsfaktor, er forholdet mellem den justerede retentionstid (volumen) af den testede komponent og den justerede retentionstid (volumen) af standarden under visse kromatografiske forhold.

Relative retentionsværdier blev brugt til at eliminere indflydelsen af visse driftsbetingelser, såsom flowhastighed og fikseringstab, på retentionsværdier.Standarden i den relative retentionsværdi kan være en komponent i den testede prøve eller en forbindelse tilsat kunstigt.
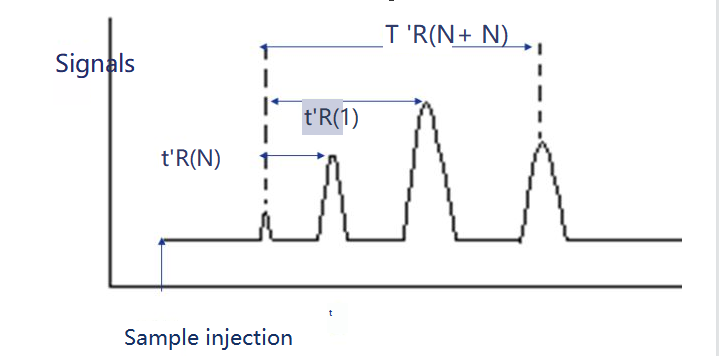
3. Fastholdelsesindeks
Retentionsindekset er retentionsindekset for stoffet i, der skal testes i en fast opløsning X. To n-alaner vælges som referencestoffer, hvoraf den ene har N kulstoftal og den anden har N+n.Deres justerede retentionstid er henholdsvis t 'r (N) og t 'r (N+n), således at den justerede retentionstid t 'r (i) for stoffet i, der skal testes, er præcis mellem dem, dvs. t'r (N).
Fastholdelsesindekset kan beregnes som følger.
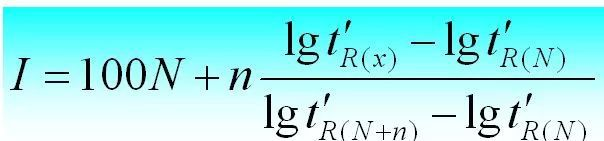
4. Kapacitetsfaktor (k)
Ved ligevægt er forholdet mellem massen af en komponent i den stationære fase (s) og den mobile fase (m), kaldet kapacitetsfaktoren.Formlen er som følger:
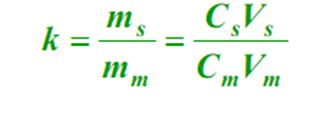
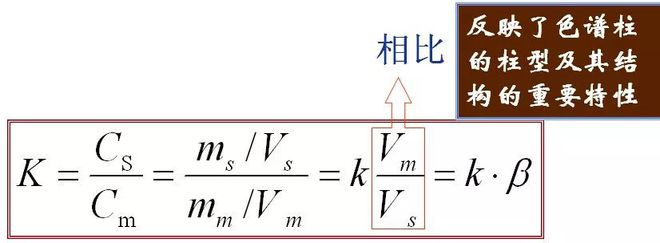
5、Fordelingskoefficient (K) I ligevægt, forholdet mellem koncentrationen af en komponent i den stationære fase(r) og den mobile fase (m), kaldet fordelingskoefficient.Formlen er som følger

Forholdet mellem K og k:
Det afspejler kolonnetypen og dens vigtige strukturegenskaber
kort opsummering
Sammenhæng mellem retentionsværdi og kapacitetsfaktor og fordelingskoefficient:
Kromatografisk adskillelse er baseret på forskellen i adsorptions- eller opløsningsevnen af hver komponent i en fast relativ prøve, som kvantitativt kan udtrykkes ved størrelsen af fordelingskoefficienten K (eller kapacitetsfaktoren k) værdi.
Komponenterne med stærk adsorptions- eller opløsningsevne har stor fordelingskoefficient (eller kapacitetsfaktor) og lang retentionstid.Omvendt har komponenterne med svag adsorption eller opløselighed en lille fordelingskoefficient og en kort retentionstid.
Grundlæggende teori om kromatografi
1. Bakketeori
(1) Fremsat - termodynamisk teori
Det startede med tårnplademodellen foreslået af Martin og Synge.
Fraktioneringskolonne: i bakken for flere gange gas-væske ligevægt, i henhold til kogepunktet for den forskellige separation.
Kolonne: Komponenterne er afbalanceret af flere partitioner mellem de to faser og adskilt i henhold til forskellige partitionskoefficienter.
(2) Hypotese
(1) Der er mange bakker i kolonnen, og komponenterne kan hurtigt nå fordelingsligevægten inden for bakkeintervallet (det vil sige højden af bakken).
(2) Den mobile fase går ind i søjlen, ikke kontinuerligt, men pulserende, det vil sige, at hver passage er et søjlevolumen.
(3) Når prøven blev tilføjet til hver søjleplade, kunne diffusionen af prøven langs søjleaksen forsømmes.
(4) Fordelingskoefficienten er ens på alle bakker, uafhængig af mængden af komponenter.Det vil sige, at fordelingskoefficienten er konstant på hver taban.
(3) Princip
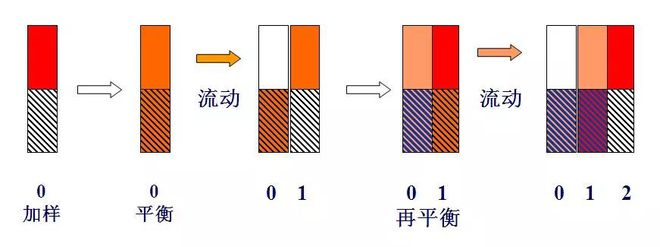
Skematisk diagram af bakketeori
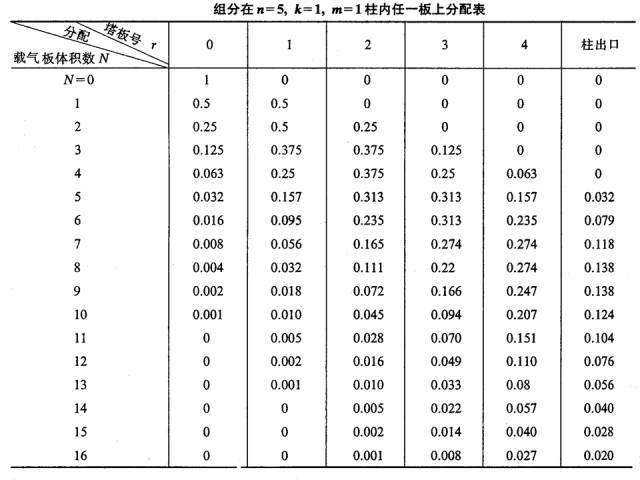
Hvis en komponent af enhedsmasse, nemlig m=1 (f.eks. 1mg eller 1μg), tilføjes til nr. 0-bakken, og efter fordelingsligevægt, fordi k=1, nemlig ns=nm, nm=ns=0,5.
Når et pladevolumen (lΔV) af bæregas kommer ind i plade 0 i form af pulsering, skubbes bæregassen indeholdende nm-komponenten i gasfasen til plade 1. På dette tidspunkt er ns-komponenten i væskefasen af plade 0 og nm-komponenten i gasfasen af plade 1 vil blive omfordelt mellem de to faser.Derfor er den samlede mængde af komponenter indeholdt i plade 0 0,5, hvori gas- og væskefaserne hver er 0,25, og den samlede mængde indeholdt i plade 1 er også 0,5.Gas- og væskefasen var også 0,25.
Denne proces gentages hver gang en ny pladevolumen bæregas pulseres ind i kolonnen (se tabel nedenfor).
(4) Kromatografisk udløbskurveligning

σ er standardafvigelsen, er retentionstiden, C er koncentrationen til enhver tid,
C, er injektionskoncentrationen, det vil sige den samlede mængde af komponenter (spidsareal A).
(5) kolonneeffektivitetsparametre
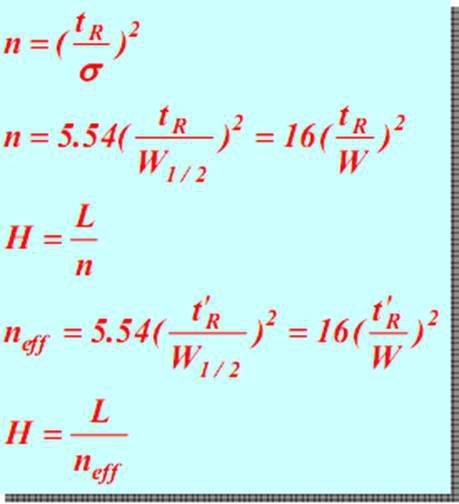
Ved en konstant tR, jo mindre W eller w 1/2 (det vil sige jo smallere top), jo større er antallet af teoretiske plader n, jo mindre er den teoretiske pladehøjde, og jo højere er søjlens adskillelseseffektivitet.Det samme gælder den effektive teoribakke neff.Derfor er det teoretiske antal bakker et indeks til at evaluere effektiviteten af kolonner.
(5) Karakteristika og mangler
> fordele
Bakketeorien er semi-empirisk og forklarer formen af udløbskurven
Komponenternes opdelings- og adskillelsesprocesser er illustreret
Et indeks til evaluering af kolonnens effektivitet foreslås
> Begrænsninger
Komponenterne kan ikke rigtig nå fordelingsligevægten i de to faser:
Langsgående diffusion af komponenter i kolonnen kan ikke ignoreres:
Indflydelsen af forskellige kinetiske faktorer på masseoverførselsprocessen blev ikke overvejet.
Forholdet mellem søjleeffekt og strømningshastighed af mobil fase kan ikke forklares:
Det er ikke klart, hvilke hovedfaktorer der påvirker kolonneeffekten
Disse problemer er tilfredsstillende løst i hastighedsteori.
2. Taksteori
I 1956 skrev den hollandske lærde VanDeemter et al.absorberede begrebet bakketeori og kombinerede de kinetiske faktorer, der påvirker bakkens højde, fremsatte den kinetiske teori om kromatografisk proces-hastighedsteori og udledte VanDeemter-ligningen.Den betragter den kromatografiske proces som en dynamisk ikke-ligevægtsproces og studerer kinetiske faktorers indflydelse på topudvidelsen (dvs. søjleeffekt).
Senere har Giddings og Snyder et al.foreslog væskekromatografi-hastighedsligningen (nemlig Giddings-ligningen) baseret på VanDeemter-ligningen (senere kaldet gaskromatografi-hastighedsligningen) og ifølge egenskabsforskellen mellem væske og gas.
(1) Van Deemter-ligning
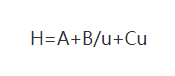
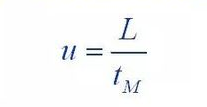
Hvor: H: er brættets højde
A: koefficient for eddy diffusion term
B: molekylær diffusionskoefficient
C: koefficient for masseoverførselsmodstandsleddet
(2) Giddings-ligning
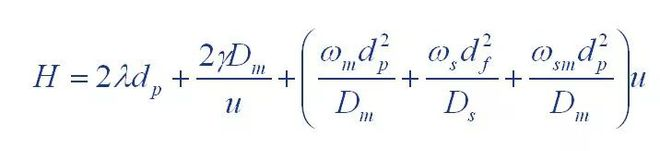
Kvantitativ og kvalitativ analyse
(1) Kvalitativ analyse
Kvalitativ kromatografisk analyse er at bestemme forbindelserne repræsenteret af hver kromatografisk top.Da forskellige stoffer har bestemte retentionsværdier under visse kromatografiske forhold, kan retentionsværdien bruges som et kvalitativt indeks.Forskellige kromatografiske kvalitative metoder er i øjeblikket baseret på retentionsværdier.
Forskellige stoffer kan dog have lignende eller identiske retentionsværdier under de samme kromatografiske forhold, det vil sige, at retentionsværdierne ikke er eksklusive.Det er således vanskeligt at karakterisere en fuldstændig ukendt prøve baseret på retentionsværdier alene.Hvis der på grundlag af forståelsen af prøvens kilde, art og formål kan foretages en foreløbig vurdering af prøvens sammensætning, og følgende metoder kan anvendes til at bestemme forbindelsen repræsenteret af den kromatografiske top.
1. Kvalitativ kontrol ved brug af rene stoffer
Under visse kromatografiske forhold har en ukendt kun en defineret retentionstid.Derfor kan det ukendte kvalitativt identificeres ved at sammenligne retentionstiden for det kendte rene stof under de samme kromatografiske forhold med retentionstiden for det ukendte stof.Hvis de to er ens, kan det ukendte stof være et kendt rent stof;Ellers er det ukendte ikke det rene stof.
Den rene stofkontrolmetode er kun anvendelig for det ukendte stof, hvis sammensætning har været kendt, hvis sammensætning er forholdsvis enkel, og hvis rene stof er kendt.
2. Relativ fastholdelsesværdimetode
Den relative retentionsværdi α refererer til justeringen mellem komponent i og referencematerialer Forholdet mellem retentionsværdier:
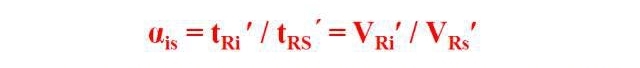
Det ændrer sig kun med ændringen af fikseringsmiddel og kolonnetemperatur og har intet at gøre med andre driftsforhold.
Ved en bestemt stationær fase og kolonnetemperatur måles de justerede retentionsværdier for henholdsvis komponent i og referencestof s og beregnes derefter i henhold til ovenstående formel.De opnåede relative retentionsværdier kan kvalitativt sammenlignes med de tilsvarende værdier i litteraturen.
3, tilsætning af kendte stoffer for at øge spidshøjdemetoden
Når der er mange komponenter i den ukendte prøve, er de opnåede kromatografiske toppe for tætte til let at kunne identificeres med ovenstående metode, eller når den ukendte prøve kun bruges til den specificerede emneanalyse.
"Først laves et kromatogram af en ukendt prøve, og derefter opnås et yderligere kromatogram ved at tilføje et kendt stof til den ukendte prøve."Komponenter med øget tophøjde kan være kendt for sådanne stoffer.
4. Bevar indeksets kvalitative metode
Retentionsindekset repræsenterer retentionsadfærden af stoffer på fiksativer og er i øjeblikket det mest udbredte og internationalt anerkendte kvalitative indeks i GC.Det har fordelene ved god reproducerbarhed, ensartet standard og lille temperaturkoefficient.
Retentionsindekset er kun relateret til egenskaberne af den stationære fase og kolonnetemperaturen, men ikke til andre eksperimentelle forhold.Dens nøjagtighed og reproducerbarhed er fremragende.Så længe kolonnetemperaturen er den samme som den stationære fase, kan litteraturværdien anvendes til identifikation, og det er ikke nødvendigt at bruge det rene materiale til sammenligning.
(2) Kvantitativ analyse
Grundlag for kromatografisk kvantificering:
Opgaven med kvantitativ analyse er at finde hundrede af komponenterne i den blandede prøve
Brøkindhold.Kromatografisk kvantificering var baseret på følgende: når driftsbetingelserne var konsistente, var
Massen (eller koncentrationen) af den målte komponent bestemmes af responssignalet fra detektoren
Det er proportionalt.Nemlig:

Grundlag for kromatografisk kvantificering:
Opgaven med kvantitativ analyse er at finde hundrede af komponenterne i den blandede prøve
Brøkindhold.Kromatografisk kvantificering var baseret på følgende: når driftsbetingelserne var konsistente, var
Massen (eller koncentrationen) af den målte komponent bestemmes af responssignalet fra detektoren
Det er proportionalt.Nemlig:
1. Metode til måling af spidsareal
Topareal er de grundlæggende kvantitative data leveret af kromatogrammer, og nøjagtigheden af toparealmåling påvirker direkte de kvantitative resultater.Forskellige målemetoder blev brugt til kromatografiske toppe med forskellige topformer.
Det er svært at finde den nøjagtige værdi af vinter i kvantitativ analyse:
På den ene side på grund af vanskeligheden ved nøjagtigt at måle det absolutte injektionsvolumen: på den anden side
Toparealet afhænger af de kromatografiske forhold, og den kromatografiske strimmel skal bibeholdes, når værdien måles
Det er hverken muligt eller bekvemt at gøre det samme.Og selvom du kan få det rigtigt
Den nøjagtige værdi, også fordi der ikke er nogen samlet standard og ikke kan anvendes direkte.
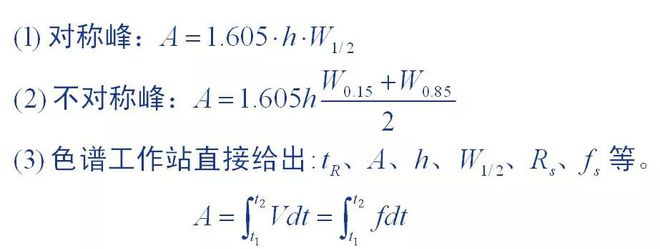
2. Kvantitativ korrektionsfaktor
Definition af kvantitativ korrektionsfaktor: mængden af komponenter, der kommer ind i detektoren (m)
Forholdet mellem dets kromatografiske topareal (A) eller tophøjde () er en proportionalitetskonstant (,
Proportionalitetskonstanten kaldes den absolutte korrektionsfaktor for komponenten.
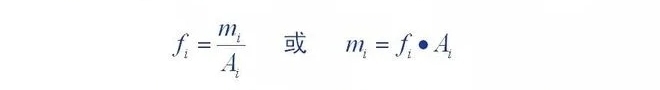
Det er svært at finde den nøjagtige værdi af vinter i kvantitativ analyse:
På den ene side på grund af vanskeligheden ved nøjagtigt at måle det absolutte injektionsvolumen: på den anden side
Toparealet afhænger af de kromatografiske forhold, og den kromatografiske strimmel skal bibeholdes, når værdien måles
Det er hverken muligt eller bekvemt at gøre det samme.Og selvom du kan få det rigtigt
Den nøjagtige værdi, også fordi der ikke er nogen samlet standard og ikke kan anvendes direkte.
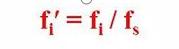
Det vil sige, at en komponents relative korrektionsfaktor er komponenten og referencematerialet s
Forholdet mellem de absolutte korrektionsfaktorer.
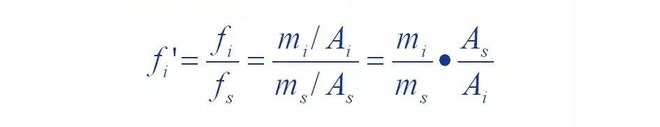
Det kan ses, at den relative korrektionsfaktor er, når kvaliteten af komponenten versus standarden.
Når stoffet s er ens, er toparealet af referencematerialet toparealet af komponenten
Mange.Hvis en komponent har masse m og topareal A, så er antallet af f'A
Værdier er lig med toparealet af referencematerialet med masse på.Med andre ord,
Gennem den relative korrektionsfaktor kan spidsarealerne af hver komponent adskilles
Omregnet til toparealet af referencematerialet lig med dets masse, derefter forholdet
Standarden er samlet.Så dette er den normaliserede metode til at finde ud af procentdelen af hver komponent
Grundlaget for mængde.
Metode til at opnå relativ korrektionsfaktor: relative korrektionsfaktorværdier blev kun sammenlignet med væren
Målingen er relateret til standarden og typen af detektor, men til operationsstrimlen
Det er lige meget.Derfor kan værdier hentes fra referencer i litteraturen.Hvis teksten
Hvis du ikke kan finde den ønskede værdi i tilbuddet, kan du også selv bestemme det.Bestemmelsesmetode
Metode: En vis mængde af det målte stof ti udvalgte referencemateriale → lavet til en bestemt koncentration
De kromatografiske topområder A og As for de to komponenter blev målt.
Det er formlen.
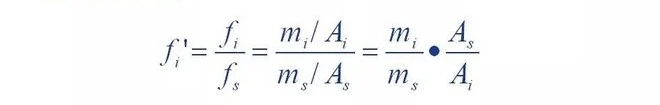
3. Kvantitativ beregningsmetode
(1) Områdenormaliseringsmetode
Summen af indholdet af alle de spidsfrie fraktioner blev beregnet som 100 % til kvantificering
Metoden kaldes normalisering.Dens beregningsformel er som følger:
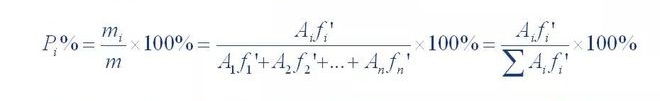
Hvor P,% er det procentvise indhold af de testede komponenter;A1, A2... A n er komponent 1. Toparealet på 1~n;f'1, f'2... f'n er den relative korrektionsfaktor for komponenterne 1 til n.
(2) ekstern standardmetode
Metoden til kvantitativ sammenligning mellem responssignalet for den komponent, der skal testes i prøven, og den rene komponent, der skal testes som kontrol.
(3) Intern standardmetode
Den såkaldte interne standardmetode er en metode, hvor en vis mængde rent stof tilsættes standardopløsningen af det testede stof og prøveopløsningen som intern standard og derefter analyseres og bestemmes.
(3) standard tilsætningsmetode
Standardtilsætningsmetoden, også kendt som den interne tilsætningsmetode, er at tilsætte en vis mængde (△C)
Referencen for teststoffet blev tilsat til prøveopløsningen, der skulle testes, og testen blev tilføjet til assayet
Toppen af prøveopløsningen efter stoffet var højere end den oprindelige prøveopløsning
Områdets stigning (△A) blev brugt til at beregne koncentrationen af stoffet i prøveopløsningen
Indhold (Cx)
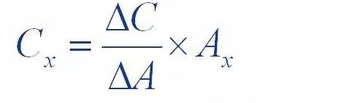
Hvor Ax er toparealet af stoffet, der skal måles i den originale prøve.

Indlægstid: 27. marts 2023